
Синдром веста что это такое
Обновлено: 04.07.2024
Синдром Веста у детей – это заболевание, крайне негативно отражающееся на жизни и здоровье ребенка. Болезнь сродни эпилепсии, но имеет куда более разрушительное влияние на здоровье ребенка.
Клиническая картина синдрома Веста у детей
Заболевание проявляется у детей в течение первого года жизни но, как правило, проходит довольно спокойно. Первые инфантильные спазмы могут происходить на фоне уже существующих нарушений психомоторной сферы. Нередко заболевание развивается у здоровых детей на фоне отсутствующих признаков. Синдром Веста у детей может развиваться без видимых проявлений и впервые показать себя в более позднем возрасте. Патология чаще наблюдается у мальчиков.
Заболевание выражается в инфантильных спазмах — внезапными сокращениями мышц ребенка. Чаще всего медики сталкиваются со смешанным видом спазмов, когда сокращаются разные группы мышц в зависимости от положения тела ребенка. Нередко во время приступов наблюдается эффект замирания: у ребенка приостанавливается дыхание, отсутствует реакция на внешние раздражители. Подобные проявления могут и не сопровождаться спазмами.
Отсутствие симметричность спазмов во время приступов свидетельствует о нарушениях в корковом слое головного мозга. Асимметричные спазмы отличаются неестественным положением головы, искажением лицевых мышц, вызывающим гримасу.
Ребенок плачет, нередко наступает дропп–атака, сопровождающаяся падением ребенка.
Этиология синдрома Веста у детей
Возникающий у ребенка синдром Веста является результатом вирусной инфекции плода в период беременности и во время родовой деятельности. Такими инфекциями может стать вирус герпеса или бактериальный менингит. К числу основных причин возникновения патологии можно отнести наличие у эмбриона гипоксии головного мозга, ишемии головного мозга, гемодинамических нарушений в результате тяжелых родов, черепно-мозговые травмы. Вирусный энцефалит, гемодинамические и хромосомные нарушения, дисплазии головного мозга могут стать причинами развития у ребенка патологии.
Диагностика синдрома Веста у детей в клинике Шнайдер
Появление у новорожденных инфантильных спазмов может вызывать подозрение на развитие эпилепсии. Очень важно отличать синдром Веста от эпилепсии. Неврологи отделения нейрохирургии клиники Шнайдер обязательно обращают внимание на другие патологии, имеющие схожесть в симптоматике с синдромом Веста. Аналогичные симптомы могут иметь синдром Сандифера, эффект кривошеи и аномальное дистоническое развитие тела ребенка.
Детальный анализ анамнеза ребенка позволит поставить правильный диагноз и начать эффективное лечение детей в Израиле. Фиксация признаков во время инфантильных спазмов дает возможность провести предварительную дифференциацию диагноза. Эти наблюдения влияют на подбор тактики лечения ребенка. Показания электроэнцефалографии дают точные данные для определения происхождения синдрома Веста у детей. Современная медицина для диагностики патологии использует традиционные методы исследования:
- Магнитно-резонансная томография обеспечивает визуализацию происходящих патологических процессов в головном мозге ребенка. Картина, имеющихся нарушений головного мозга, наличие других патологий позволяет получить точное представление о развитии патологии. Преимущество данной методики исследование заключается в безопасности для детского организма;
- Рентгенологическое исследование и компьютерная томография в процессе исследования новорожденных может нанести вред ребенку;
- Проведение разносторонних исследований, включая систематическое наблюдение за ребенком и изучение анамнеза, позволит избежать ошибки при постановке диагноза.
Лечение синдрома Веста у детей в Израиле
В отличие от эпилепсии, которая достаточно успешно лечиться в отделении детской нейрохирургии клиники Шнайдер, синдром Веста отличается худшим прогнозом. Происходящие в результате инфантильных припадков серьезные изменения психомоторной деятельности, нарушения функций головного мозга приводят к необратимым процессам.
Основной метод лечения детей в Израиле патологии подбирается индивидуально. Чаще всего используются медикаментозные средства. Основная задача медикаментозного лечения для детей с диагнозом синдром Веста у детей — противосудорожная терапия. Снижение частоты инфантильных спазмов, уменьшение длительности приступов позволяет улучшить состояние ребенка и притормозить негативные изменения в детском организме. Применение стероидных и гормональных препаратов обеспечивает восстановление функций головного мозга, снижает активность импульсов головного мозга. Витаминотерапия дает возможность ребенку спокойно пережить состояние, наступающей после судорожных припадков.
Лечение синдрома Веста у детей в Израиле в клинике Шнайдер
Главный результат лечения детей в Израиле в отделении деткой нейрохирургии клиники Шнайдер заключается в существенном снижении частоты и интенсивности приступов. Правильный подбор медикаментов, их дозировки обеспечивают нормальное развитие ребенка, а также помогают устранить нарушения психомоторной деятельности детского организма. Специалисты отделения детской нейрохирургии клиники Шнайдер отмечают, что дети по-разному реагируют на терапию, поэтому очень важно проводить тщательную диагностику. Индивидуально подобранное лечение поможет побороть болезнь и обеспечить ребенку нормальное качество жизни.

Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
Синдром Веста - синдром тяжелой эпилепсии, состоящей из триады признаков: инфантильных спазмов, характерными изменениями в межприступный период электроэнцефалограммы (hypsarrhythmia) и умственной отсталости. Болезнь получила свое название благодаря британскому доктору Весту, который впервые описал все его симптомы в 1841 году, наблюдая за своим больным сыном. Также это заболевание известно под такими названиями: гипсаритмия Гибса, поклонные судороги, спазм или тик Салаама, миоклоническая энцефалопатия с гипсаритмией.
Код по МКБ-10
Эпидемиология

[1], [2], [3], [4], [5], [6], [7]
Причины синдрома Веста
Его можно отнести к группе так называемых энцефалопатических эпилепсий, когда приступы возникают на фоне невоспалительных заболеваний головного мозга.
Данная болезнь чаще всего встречается в раннем детском возрасте и имеет множество причин:
- Врожденная патология в развитии головного мозга (к примеру, туберозный склероз).
- Генетические болезни, генные мутации и расстройства метаболизма.
- Инфекции головного мозга.
- Родовые внутричерепные кровоизлияния, гипоксия мозга (особенно у недоношенных детей).
- Асфиксия.
Именно последняя причина считается самой распространенной при развитии этого опасного синдрома. Асфиксия плода часто развивается вследствие осложненных родов.
Обсуждается роль генетических дефектов в этиологии Синдрома Веста. Идентифицировано два генетических дефекта. Первый - мутация в коротком плече X хромосомы. Ген ARX ассоциируется с ранним началом инфантильных спазмов. Второй - дефект в циклин-зависимой киназе и белке 5 (CDKL5).
Иногда определить, что именно повлияло на проявление синдрома довольно сложно. Врачи, в таких случаях, ставят диагноз идиопатический синдром. Тех пациентов, у которых нет признаков заболевания, но имеются изменения в ЭЭГ (или наоборот) относят к 1 группе риска. Им не нужно специальное лечение, но они обязаны ежегодно проходить обследование.
Если есть основные признаки и изменения в ЭЭГ, то больных относят к 2 группе риска. Им назначают определенное лечение. Также они проходят два раза в год обследование. Последняя группа риска – это те, кто отличается ярко выраженными симптомами и изменениями в ЭЭГ.
[8], [9], [10], [11]
Факторы риска
Пренатальные расстройства, связанные с инфантильными спазмами, следующие:
- Гидроцефалия.
- Микроцефалия.
- Гидроанэнцефалия.
- Шизэнцефалия.
- Полимикрогирия.
- Синдром Штурге-Вебера.
- Клубневый склероз.
- Трисомия по 21 хромосоме.
- Гипоксически-ишемическая энцефалопатия.
- Врожденные инфекции.
- Травмы.
Перинатальные нарушения, приводящие к развитию синдрома Веста, следующие:
- Гипоксически-ишемическая энцефалопатия.
- Менингит.
- Энцефалит.
- Травмы.
- Внутричерепные кровоизлияния.
[12], [13], [14], [15], [16], [17]
Патогенез
Инфантильные спазмы, как полагают, возникают вследствие аномальных взаимодействий между корой и стволовых структур мозга. В патогенезе главным является незрелость центральной нервной системы и нарушение обратной связи оси "мозг-надпочечники". Под влиянием различных факторов стресса в незрелом мозгу производит ненормальное, чрезмерное выделение кортикотропин-рилизинг-гормона (КРГ), в результате чего и возникают спазмы.
[18], [19], [20], [21], [22], [23], [24], [25]
Симптомы синдрома Веста
Среди симптомов этого заболевания следует выделить:
- Частые припадки эпилептического характера. Вылечить их достаточно сложно.
- Характерные для этой болезни изменения в ЭЭГ (гипсаритмия).
- Отчетливые нарушения в психомоторном развитии.
Почти у всех детей с синдромом Веста (90%) симптомы проявляются практически сразу после рождения (от 4 до 8 месяцев). Припадки отличаются небольшой длительностью, поэтому поставить правильный диагноз получается не сразу.
Синдром Веста считается тяжелым заболеванием, которое угрожает жизни больного. Его также называют инфальтильными спазмами.
Приступ происходит всегда одинаково: все тело ребенка наклоняется резко вперед, а голова сильно сгибается. Такие судороги обычно возникают в момент пробуждения младенца или во время засыпания. При этом приступов может быть от десяти до сотни за один день.
Часто во время приступов ребенок может терять сознание. Из-за них у младенцев происходит отставание в развитии психомоторики. Дети с синдромом Веста плохо реагируют на окружающую среду, практически не идут на контакт с родственниками.
Первые признаки
Первым признаком приступа синдрома Веста является сильный плач ребенка, поэтому часто врачи ставят таким малышам диагноз колики. Стандартными признаками данного заболевания можно назвать:
- Сильные наклоны всего туловища вперед.
- Генерализованные судороги в нижних и верхних конечностях, всем туловище.
- Конечности непроизвольно разводятся в стороны.
Обычно такой приступ длиться не больше одной-двух секунд. Наступает короткая пауза и приступ возобновляется снова. В некоторых случаях спазмы являются единичными, но чаще всего они идут чередой.
Дети с синдромом Веста часто сильно раздражительные, отстают в развитии, разной степени тяжести. Груднички с этим заболеваниям ведут себя часто, как слепые.
[26], [27]
Эпилепсия при синдроме Веста
Синдром Веста является одним из вариантов генерализированной эпилепсии катастрофического характера. Он может быть симптоматическим (в большинстве случаев) или криптогенным (только до 10% от всех случаев). Классический вариант синдрома Веста можно охарактеризовать ярко выраженными салаамовыми или миоклоническими спазмами. Иногда спазмы имеют форму коротких серийных кивков головы.
Эпилепсия при синдроме Веста развивается по причине различных неврологических патологий или же без определенных нарушений в работе центральной нервной системы. Инфантильные спазмы приводят к замедленному развитию психических и моторных функции ребенка, что в будущем может стать причиной выраженного отставания в общем развитии.
В 80% случаев у детей с синдромом Веста наблюдается детский церебральный паралич, микроцефалия, атонические и атактические нарушения.
[28], [29], [30]
Энцефалопатия при синдроме Веста
Как уже говорилось выше, синдром Веста также известен под названием миоклоническая энцефалопатия с гипсаритмией. Гипсаритмия является типичным, но не патогномоничным паттерном энцефалограммы у больных на это заболевание.
Стандартная гипсаритмия характеризируется непрерывной аритмичной и высокоамплитудной медленноволновой активностью, а также имеет многочисленные спайки и острые волны. При этом между разными отделами гемисфер не существует синхронизации. Иногда паттерны могут отличаться амплитудной ассиметрией.
Гипсаритмия почти полностью замещает собой основную фоновую активность.
[31], [32], [33], [34], [35]
Симптоматический синдром Веста
Как правило, в 75% случаев, начинается синдром Веста во время второго или третьего квартала жизни младенца. Первые месяцы развитие ребенка кажется вполне нормальным и только потом появляются судороги, которые являются патогномоничным первым признаком. Иногда у пациентов наблюдается задержка психомоторного развития. Очень редко можно увидеть изменения в ЭЭГ.
Миоклонии или мышечные судороги затрагивают практически все тело. Во время таких припадков тело и конечности малыша сгибаются. Судороги и сокращения в сгибательных мышцах могут быть двусторонними, синхронными, внезапными, симметричными и длятся максимум 10 секунд. Иногда они повторяются до ста раз в сутки.
В некоторых случаях конвульсионный приступ может затрагивать только какую-то одну группу мышц. Нижние и верхние конечности во время судорог разбрасывает в стороны, голова сгибается и ложится на грудную клетку. Если частота приступов высокая, ребенок может впадать в сон.
На сегодняшний день существует три отдельных варианта синдрома Веста, которые отличаются друг от друга степенью и характером поражения мышц:
У детей с синдромом Веста сразу же после рождения или по истечении шести месяцев проявляется отставание в моторном и психическом развитии. Частые судороги только усугубляют ситуацию.
Мозжечковый синдром при синдроме Веста
В некоторых случаях при синдроме Веста может возникать мозжечковый синдром. Это поражение мозжечка или же нарушение его связей с другими отделами головного мозга. Основные признаки мозжечкового синдрома:
- Интенционное дрожание пальцев рук (особенно во время движения).
- Адиодохокинез.
- Вялость и дряблость мышц.
- Появляется симптом отсутствия обратного толчка.
- Системные головокружения.
[36], [37]
Осложнения и последствия
Течение синдрома Веста довольно тяжелое практически во всех случаях, так как он проявляется серьезными нарушениями мозга. Очень редко данное заболевание может проходить с помощью консервативного лечения. Но обычно даже после эффективной терапии с течением времени появляются рецидивы.
Практически всегда после выздоровления пациента у него отмечают серьезные и довольно тяжелые неврологические остаточные явления: эпилепсия и ее эквиваленты, экстрапирамидные проявления. Также у больных проявляются психические нарушения: идиотизм или легкое слабоумие.
Только в 2% случаю (по Гиббсу) происходит спонтанное полное выздоровление.
[38], [39], [40], [41], [42], [43], [44], [45]
Диагностика синдрома Веста
Диагностика синдрома Веста осуществляется с помощью таких врачей: нейрохирурга, эпилептолога, невролога, педиатра, иммунолога, эндоскописта и эндокринолога. Благодаря использованию современных аппаратов можно поставить более точный диагноз. Обычно используются: радиомагнитная и компьютерная томография, краниоскопия (в очень редких случаях), цереброангиография. Также для того, чтобы выявить патологический очаг эпилептических приступов проводят нейрофизиологические обследования.
Самыми популярными методами диагностики синдрома Веста являются: электроэнцефалография и газовая энцефалография.
Благодаря электроэнцефалографии можно выявить гипсаритмию биологических кривых:
В некоторых случаях с помощью газовой энцефалографии можно увидеть расширение желудочков головного мозга. На поздних стадиях синдрома Веста отмечается гидроцефалия.
[46], [47], [48], [49], [50], [51], [52]
Дифференциальная диагностика
Дифференцировать синдром Веста можно как с неэпилептическими заболеваниями, которые являются частыми в младенческом возрасте (колики, двигательное беспокойство, инфантильная мастурбация, гиперэксплексия, респираторный приступ), так и с некоторыми эпилептическими синдромами (например, фокальная эпилепсия). Очень важную роль в дифференциальной диагностике играет электроэнцефалография.
[53]
К кому обратиться?
Лечение синдрома Веста
Лечение индивидуальное в каждом конкретном случае и зависит от причины, вызвавшей синдром Веста и состояния развития мозга.
Основным методом лечения синдрома Веста на сегодняшний день является стероидной терапии с адренокортикотропным гормоном (АКТГ) (Sabril, вигабатрин). Но такое лечение должно быть крайне осторожным и проходит под строгим наблюдением врача, так как и стероидные препараты и вигабатрин имеют множество серьезных побочных действий. Также необходимо подобрать подходящие противосудорожные средства, а также лекарственные препараты, которые помогут нормализировать кровоснабжение мозга.
Иногда нейрохирург должен провести операцию, в ходе которой рассекаются спайки мозговой оболочки и удаляется патологический очаг с врожденными аневризмами сосудов. Эта процедура проходит посредством стереотаксической хирургии и различных эндоскопических способов. Новым и довольно дорогим методом терапии синдрома Веста является использование стволовых клеток. Он считается эффективным, но непопулярным из-за высокой стоимости процедуры.
Суть этого метода заключается в том, что нарушенный участок мозга восстанавливается с помощью стволовых клеток.
Идиопатическая форма синдрома Веста обычно лечится с помощью специальных препаратов:
Терапия является эффективной, если снижается количество и частота припадков. При правильно подобранной терапии в будущем ребенок будет нормально развиваться и обучаться.
Но стоит понимать, что даже современные препараты несут множество побочных действий:
- Нарушение концентрации.
- Усталость.
- Аллергические реакции кожи.
- Депрессии.
- Нервные поражения.
- Печеночная недостаточность.
ЛФК при синдроме Веста
Лечебная физкультура при синдроме Веста должна проводиться под строгим наблюдением реабилитолога и врача спортивной медицины, чтобы не усугубить приступы судорог. Этот метод терапии является довольно популярным, но не дает эффективных результатов без комплекса медикаментозных средств.
Случаи излечения
Отсутствие приступов длительное время при синдроме Веста не может говорить о том, что заболевание перешло в стадию ремиссии. Но некоторые врачи считают, что если спазмы, судороги, гипсаритмия и изменения в ЭЭГ не были замечены в течение месяца, то это можно считать выздоровлением. К сожалению, на сегодняшний день такие случаи довольно редкие. По некоторым источникам, только 8% всех пациентов излечиваются полностью, по Гиббсу это количество достигает всего лишь 2%.
[54], [55], [56]
Брагина О.Н. 1 Иванова Е.А. 1 Антонова Е.С. 1 Филиппов И.Ю. 1 Сергеева Т.А. 1 Максимов А.Г. 1 Гурьянова Е.А. 1

1. Fischer R.S., Acevedo C., Arzimanoglou A., Bogacz A., Cross J.H., Elger C.E., Engel J.Jr, Forsgren L., French J.A., Glynn M., Hesdorffer D.C., Lee B.I., Matshern G.W., Mosche S.L., Perucca E., Scheffer I.E., Tomson T., Watanabe M., Wiebe S. ILAE official report: a practical clinical definition of epilepsy. Epilepsia. 2014. V. 55(4). Р. 475-482. DOI: 10.1111/epi.12550.
2. Мухин К.Ю., Миронов М.Б. Эпилептические спазмы // Русский журнал детской неврологии. 2014. Т.9. №4. С. 9-20.
3. Sankar R., Koh S., Wu J, Menkes J.H. Paroxysmal disorders. In: Menkes J.H., Sarnat H.B., Maria B.L. Child Neurology. 7th ed. Philadelphia, PA: Lippincott, Williams, and Wilkins, 2006. 857-942.
4. Pavone P., Striano P., Falsaperla R., Pavone L., Ruggieri M.. Management of infantile spasms. Transl Pediatr. 2015. Vol. 4. №4. P. 70-260. DOI: 10.3978/j.issn.2224-4336.2015.09.01.
5. Wirrell E.C., Shellhaas R.A., Joshi C., Keator C., Kumar S., Wendy G. How should children with West syndrome be efficiently and accurately investigated? Results from the National Infantile Spasms Consortium. Mitchell, and Pediatric Epilepsy Research Consortium (PERC) Epilepsia. 2015. Vol. 56. № 4. P. 617-625. DOI: 10.1111/epi.12951.
6. Paciorkowski A.R., Thio L.l., Dobyns W.B. A genetic and biologic classification of infantile spasms. Pediatr Neurol. 2011. Vol. 45. №6. P. 355–67. DOI: 10.1016/j.pediatrneurol.2011.08.010.
7. Nash K., Sullivan J. Myoclonic seizures and infantile spasms. In Swaiman K.F., Ashwal S., Ferriero D.M., Schor N.F. editors. Swaiman’s pediatric Neurology: Principles and Practice. 5th ed. Edinburgh: Elsevier Saunders. 2012. P. 774-789.
8. Sanmaneechai O., Sogawa Y., Silver W., Ballaban-Gil K., Moshe S.L., Shinnar S. Treatment outcomes of West syndrome in infants with Down syndrome. Pediatr Neurol. 2013. Vol. 48. № 1. P. 42-47.
9. Stafstrom C.E., Konkol R.J. Infantile spasms in children with Down syndrome. Dev Med Child Neural. 1994. Vol. 36. P. 576-585.
10. Эпилепсия: синдром Веста (эпилептические спазмы). Клинические рекомендации. Санкт-Петербург: 2018. 72 с.
11. Riikonen R. Recent advances in the pharmacotherapy of infantile spasms. CNS Drugs. 2014. Vol. 28. № 4. P. 279-90.
12. Knupp K.G. Hormonal therapy with vigabatrin is superior to hormonal therapy alone in infantile spasms. J. Pediatr. 2017. Vol. 184. P. 235-238. DOI: 10.1016/j.jpeds.2017.02.051.
13. Song J. M., Hahn J., Kim S. H., Chang M. J. Efficacy of Treatments for Infantile Spasms: A Systematic Review. Clin. Neuropharmacol. 2017. Vol. 40. №2. P. 63-84. DOI: 10.1097/ WNF.0000000000000200.
14. Iype M., Saradakutty G., Puthuvathra Abdul Mohammed Kunju. Infantile spasms: A prognostic evaluation. Ann. Indian Acad. Neurol. 2016. Vol. 19. №2. P. 228–235. DOI: 10.4103/0972-2327.173314.
Синдром Веста (СВ) – это тяжелый эпилептический синдром, состоящий из триады инфантильных спазмов, интериктальной электроэнцефалограммы (ЭЭГ), называемой гипсаритмией, и регрессом или задержкой психомоторного развития.
Согласно данным ILAE (International League Against Epilepsy, Международная противоэпилептическая лига) в общей детской популяции СВ встречается достаточно редко: 1 на 2000 новорожденных. Чаще встречается у лиц мужского пола (60%) [1].
Инфантильные спазмы (ИС) – быстрые сокращения мышц продолжительностью 1–2 секунды, занимающие по своей скорости промежуточное положение между миоклоническими и тоническими сокращениями у ребенка в возрасте до 1 года [2].
ИС включают в себя внезапные, двусторонние и симметричные сокращения мышц шеи, туловища и конечностей. Они могут быть классифицированы на очаговые и диффузные. Тип припадка зависит от того, какие группы мышц (сгибатели или разгибатели) подвергаются преимущественному воздействию, и от степени сокращения. Сгибательные спазмы (42% случаев) и смешанные сгибательно-разгибательные спазмы (50% случаев) считаются наиболее характерным для ИС типом судорог. Приступ может быть кратковременным в виде внезапной остановки активности ребенка или подергивания глазных яблок, может маскироваться под рефлекс Моро. Судороги могут быть одиночными, но чаще объединяются в серии и связаны с засыпанием или пробуждением ребенка. Во время или после серии инфантильных спазмов отмечаются раздражительность и плач младенца.
Наиболее часто ИС развиваются в возрасте от 3 до 8 месяцев, и только в 8% случаев впервые встречаются у детей старше 1 года [3]. При оценке нервно-психического развития ребенка с ИС обращает на себя внимание временная связь между появлениями спазмов и задержкой или регрессом психомоторного развития. Данная связь не всегда проявляется, если у ребенка уже имеется задержка в развитии.
В 60–70% всех случаев этиологию заболевания позволяет определить магнитно-резонансная томография (МРТ) головного мозга, где можно увидеть наличие различных повреждений ткани головного мозга [4, 5].
Крайне важно установить этиологию инфантильных спазмов, что позволит подобрать эффективную терапию и улучшить прогноз. Авторы выделяют две группы пациентов с СВ: с криптогенными и симптоматическими спазмами. Криптогенные спазмы составляют меньшинство случаев ИС (10–40%). В симптоматических ИС, которые имеют место у 60–70% пациентов, регистрируются различные пренатальные, перинатальные и постнатальные нарушения [6]. Пренатальная этиология включает пороки развития центральной нервной системы, хромосомные аномалии, моногенные генетические нарушения, нейрокутанный синдром, врожденные инфекции центральной нервной системы (TORCH). Перинатальные повреждения включают гипоксическую ишемическую энцефалопатию, травмы. К постнатальным факторам относятся внутричерепные инфекции, опухоли головного мозга и др. [7].
Важно отметить, что около 30–40% всех СВ являются генетически детерминированными энцефалопатиями. Описано частое возникновение ИС у детей с туберозным склерозом и синдромом Дауна. По данным литературы, 6% пациентов с синдромом Веста страдают трисомией по 21 хромосоме, что имеет большое клиническое значение [8, 9].
Цель: представленный клинический случай предназначен продемонстрировать сложности подбора эффективной противоэпилептической и гормональной терапии синдрома Веста на фоне тяжелой сопутствующей патологии.
Больная Г. 6 месяцев с диагнозом Q90.0 Синдром Дауна. Трисомия 21, мейотическое нерасхождение. Беременность наступила методом экстракорпорального оплодотворения, отягощена возрастом матери (35 лет). На 16-й неделе проведена пренатальная диагностика, которая выявила синдром Дауна. Родителями принято решение сохранить беременность.
По данным профилактических осмотров участкового врача-педиатра установлено, что ребенок рос и развивался с задержкой развития – переворачивается с 5 месяцев, не сидит, не ползает, не упирается на ножки, на предметах фиксирует взгляд кратковременно. Также отмечается регресс приобретенных навыков – перестала тянуться к игрушкам и брать их в руки. Заболевание началось 12.01.2020 в 16:00 часов с серийных сгибательно-разгибательных спазмов верхних и нижних конечностей в виде раскидывания в стороны. Спазмы двусторонние, симметричные, длительностью до 3–5 минут, сопровождаются вегетативной симптоматикой, возникают после пробуждения, после приступа ребенок вялый и засыпает. Аналогичный приступ случился 13.01.2020 в 07:00 часов, 14.01.2020 в 10:30 и 17:00, длительность приступа до 7 минут. 15.01.2020 после пробуждения приступ повторился в 6:00 и 14:20 часов. Госпитализированы на лечение в психоневрологическое отделение.
Наследственность по заболеваниям нервной системы не отягощена.
Состояние средней степени тяжести, обусловленное поражением центральной нервной системы, частотой и интенсивностью эпилептических приступов, задержкой в нервно-психическом развитии.
Физическое развитие микросоматическое дисгармоничное, нормотрофия. Температура 36,4?С. Кожные покровы чистые, бледно-розовые. Видимые слизистые нормальной физиологической окраски, в зеве спокойно. Лимфатические узлы не увеличены. Обращает на себя внимание диффузная грубая мышечная гипотония в конечностях. Костно-суставная система без особенностей. Органы дыхания: ЧДД 35 в минуту, перкуторно слышен ясный легочной звук, аускультативно – дыхание пуэрильное, проводится по всем полям, хрипы не выслушиваются. Оценка сердечно-сосудистой системы: ЧСС 120 в минуту, тоны сердца ритмичные, ясные. Живот при пальпации мягкий, безболезненный. Печень выступает на 1,5 см от края реберной дуги, селезенка не пальпируется. Стул ежедневный, диурез в норме.
В неврологическом статусе: сознание ясное, поведение адекватное, малоактивное. Менингеальные симптомы отрицательные. Череп округлой формы, уплощенный затылок, голову удерживает слабо. Окружность головы 41 см, окружность груди 40,5 см. Размеры большого родничка 2,5 х 2,0 см. Черепно-мозговая иннервация: лицо симметрично, плоская переносица, монголоидный разрез глаз, глазные щели d=s, зрачки равной величины, живо реагируют на свет, за предметами следит, при взгляде вверх фиксация кратковременная. Язык по средней линии, глотание и фонация не нарушены. Движения в конечностях в достаточном объеме. Мышечный тонус – диффузная грубая гипотония. Опора на ножки слабая, шаговых движений нет. Рефлексы со слизистых: положительные корнеальные, подошвенные, глоточный рефлексы, брюшные d=s. Сухожильные рефлексы живые, равные.
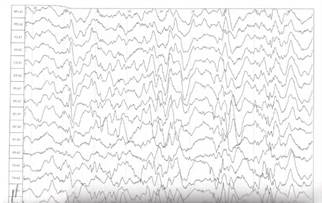
Рис. 1. ЭЭГ до лечения: картина модифицированной гипсаритмии с персистирующим супрессивно-взрывным паттерном
На МРТ головного мозга признаков патологических изменений вещества головного мозга не было выявлено. При проведении нейросонографии обнаружены эхопризнаки псевдокисты сосудистого сплетения левого бокового желудочка. На УЗИ органов брюшной полости эхопатологии не выявлено. На ЭХО-КГ сердца створки и полости не изменены, сократительная способность миокарда сохранена.
Целями лечения синдрома Веста являются быстрое купирование спазмов, нормализация ЭЭГ и улучшение психического развития ребенка. Пациентам с СВ рекомендованы гормональная терапия, антиэпилептические препараты и препараты метаболического действия [11]. Препаратами выбора служат гормональные препараты и противоэпилептический препарат Вигабатрин [12]. Однако, учитывая высокую стоимость и труднодоступность Вигабатрина, специалисты применяют альтернативные противоэпилептические препараты, эффективность которых оценивается от 40% до полного купирования приступов. К ним относятся: вальпроевая кислота, Топирамат, Леветирацетам, Зонисамид [13]. На начальном этапе лечения ребенку был назначен препарат вальпроевой кислоты в дозе 30 мг два раза в сутки. Данный препарат применялся в течение трех дней (16.01.2020 – 18.01.2020) без должного эффекта – частота приступов увеличилась. 16.01.2020 отмечались спазмы каждые 3–4 часа длительностью каждого эпизода по 4–5 минут, они сопровождались сгибательно-разгибательными спазмами конечностей и эмоциональной возбудимостью ребенка. 17.01.2020 частота приступов также 6 раз в день, длительность приступов – 4–10 минут. 18.01.2020 приступы возникали 6 раз в день, длительность – 5–10 минут с генерализацией процесса на туловище и конечности. Далее доза препарата вальпроевой кислоты была увеличена – 30 мг в утреннее время, 60 мг в вечернее. На данной дозировке препарата улучшений не отмечалось, что привело к решению о подключении гормональной терапии. Ребенку проведены пульс-терапия метилпреднизолоном, внутривенное капельное введение препарата в дозе 30 мг/кг/сутки в течение 3 дней. С первых суток применения пульс-терапии отмечается улучшение состояния ребенка – 20.01.2020 частота приступов снизилась до 4 раз в сутки, 21.01.2020 – 3 раза в сутки, 22.01.2020 – приступы купировались.
Далее пациент переведен на таблетированную форму метилпреднизолона в поддерживающей дозе 14 мг/сутки (8 мг в 6:00 ч, 4 мг в 9:00 ч, 2 мг в 12:00 ч) в течение 7 дней.
После купирования приступов пациенту проведена контрольная ЭЭГ с депривацией сна, которая показала положительную динамику: физиологические паттерны сна дифференцированы, эпиактивность не регистрируется (рис. 2).
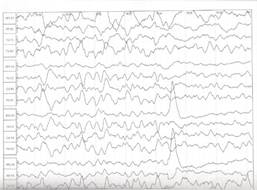
Рис. 2. ЭЭГ с положительной динамикой после проведенного лечения
По данным литературы, 6% пациентов с синдромом Веста страдают синдромом Дауна, что значительно затрудняет диагностику синдрома Веста и, соответственно, своевременное начало терапии [8]. Трудности в диагностике создает определение задержки психоневрологического развития, которая обычно является предвестником инфантильных спазмов, но сопровождает и синдром Дауна. Следовательно, заподозрить СВ мы можем только со старта инфантильных спазмов или после целенаправленного проведения ЭЭГ, как и произошло в данном случае.
В приведенном клиническом случае выявлено три этиологических фактора для развития синдрома Веста: беременность получена путем экстракорпорального оплодотворения, развитие трисомии по 21 хромосоме, осложненное течение пре- и интранатального периодов. Беременность протекала под угрозой невынашивания в I и II половине беременности, что создало условия для гипоксически-ишемической травмы плода. Также плод находился в ягодичном предлежании, что стало причиной оперативного родоразрешения.
Терапия впервые выявленного синдрома Веста трудна, потому что требует индивидуального подхода к каждому пациенту. В аспекте выбора базисной терапии данного пациента мы видим обоснованную тактику: стартовым препаратом стала вальпроевая кислота, подбор дозы лекарственного средства осуществлялся ступенчато. На начальном этапе терапии пациент не отвечал на монотерапию противосудорожным препаратом вальпроевой кислоты, что привело к решению увеличить дозу. Это не дало положительного эффекта. Затем на основании клинических рекомендаций был введен гормональный препарат. В течение трех дней проведена пульс-терапия метилпреднизолоном (внутривенное капельное введение препарата в дозе 30 мг/кг/сутки в течение 3 дней), которая с первых суток дала положительный эффект – купировались приступы ИС, затем ребенок переведен на поддерживающую дозу метилпреднизолона с пероральным приемом в дозе 14 мг/сутки в течение 7 дней.
Диспансерное наблюдение за пациентами с синдромом Веста осуществляется врачом-неврологом по месту жительства пациента. Кратность визитов определяется индивидуально врачом-неврологом. Если возникает рецидив приступа, то пациент должен быть направлен к врачу-эпилептологу. Показано проведение ЭЭГ через 1 месяц после выписки из психоневрологического отделения, затем 1 раз в 6 месяцев для контроля эпилептиформных разрядов. Длительность подобного ЭЭГ контроля не определена, так как прогноз дальнейшей эволюции эпилепсии у пациентов разный [14]. Для осуществления наглядного контроля за течением заболевания родителям пациента рекомендовано ведение дневника приступов.
Профилактика ИС проводится путем приема пероральной формы метилпреднизолона в течение 3 месяцев и регулярного приема препарата вальпроевой кислоты. В большинство национальных рекомендаций также внесена необходимость оценки развития ребенка, перенесшего синдром Веста. Минимальная кратность такой оценки – 2 раза: в возрасте около 1,5 лет и перед школой. Для оценки развития могут быть использованы различные шкалы (Бейли, DP3, Гриффитс и др.) [10].
В последние годы достигнуты огромные успехи в лечении синдрома Веста. Медикаментозная ремиссия отмечается, по данным разных авторов, у 72% больных. Однако, несмотря на успехи, достигнутые в лечении синдрома Веста, его общий прогноз остается серьезным. Согласно наблюдениям, описанным в зарубежной литературе, у половины больных с инфантильными спазмами к подростковому возрасту произошла трансформация в различные фокальные или мультифокальные формы эпилепсии. Умственная отсталость наблюдается у подавляющего большинства пациентов с синдромом Веста, составляя 90% в исследовании R. Riikonen. У половины больных констатируется тяжелая умственная отсталость. При криптогенных формах умственное развитие пациентов страдает в меньшей степени, но в большинстве случаев отмечаются выраженные когнитивные нарушения. Наиболее значимым фактором, определяющим прогноз интеллектуального развития, является этиология. Дети с криптогенными ИС гораздо чаще (38–78%) имеют нормальный или близкий к нормальному интеллект по сравнению с детьми с симптоматическими спазмами (2–18%). В то же время при синдроме Дауна возможен относительно благоприятный исход течения симптоматических инфантильных спазмов. Признаки благоприятного исхода СВ — нормальный неврологический статус и нормальное психомоторное развитие до момента возникновения спазмов, изначальное отсутствие иных видов судорог, более старший возраст дебюта инфантильных спазмов, короткая длительность приступов и рано начатая эффективная терапия. В данном клиническом примере можно надеяться на благоприятный исход, так как диагноз был поставлен своевременно и рано начата терапия, которая дала положительный результат.

Актуальность. Синдром Веста — младенческая эпилептическая энцефалопатия, характеризующаяся триадой симптомов: инфантильные спазмы, изменения на электроэнцефалограмме (ЭЭГ) в виде гипсаритмии и задержка психомоторного развития. Определение предикторов исхода заболевания даст возможность быстро подобрать оптимальную терапию, определить сроки динамического наблюдения и улучшить результаты лечения.
Цель работы — выявить предикторы исхода синдрома Веста.
Материалы и методы. В исследование вошли 132 пациента, которые проходили лечение с 2000 по 2018 г. Возраст детей на момент начала наблюдения составлял 5 мес. – 17 лет 11 мес. Возраст дебюта спазмов варьировал от 1-х суток жизни до 3 лет 2 мес. По этиологическому критерию пациентов разделили на три группы: 1-я группа — пациенты со структурной формой (60 пациентов; 45,5 %), 2-я группа — пациенты с генетической формой (39 детей; 29,5 %), 3-я группа — пациенты с невыясненной этиологией заболевания (33 ребенка; 25,0 %). При оценке терапии внимание уделяли эффективности препаратов первых трех линий, проводимой гормональной терапии, а также дальнейшему подбору антиэпилептических препаратов.
Результаты. Эпилептические спазмы купированы у 76 (57,6 %), все приступы — у 48 (36,4 %) детей. У пациентов 3-й группы отмечена бóльшая частота купирования спазмов (87,9 % против 48,7 и 46,7 % соответственно) и полной ремиссии (72,7 против 26,7 и 27,6 % соответственно), чем в 1-й и 2-й группах. Позитивное прогностическое значение для купирования спазмов имели: нормальное нервно-психическое развитие до дебюта спазмов, отсутствие эпиактивности или наличие региональной эпиактивности на ЭЭГ в динамике, наличие диффузных изменений на МРТ. Негативное прогностическое значение имели: неонатальные судороги, наличие эпиактивности на ЭЭГ и очагового дефицита до спазмов, наличие других приступов, кроме спазмов, патология зрения и слуха, необходимость применения ≥2 препаратов. У пациентов, достигших ремиссии, отмечена лучшая компенсация моторного и психоречевого развития.
Выводы. Предикторами неблагоприятного исхода синдрома Веста можно считать: структурную и генетическую форму заболевания, неонатальные судороги, наличие эпилептиформной активности (ЭА) на ЭЭГ, нарушение нервно-психического развития и наличие очаговой патологии до спазмов, наличие других приступов кроме спазмов, сохранение ЭА в динамике, неэффективность терапии первой линии.
Ключевые слова
Полный текст
Актуальность
Синдром Веста является одной из катастрофических эпилепсий детского возраста вследствие сложности контроля приступов и задержки умственного развития [14]. Впервые данное заболевание было описано английским педиатром W.J. West в 1841 г. в журнале Lancet на примере болезни собственного сына [2, 3]. Синдром Веста — младенческая эпилептическая энцефалопатия, характеризующаяся триадой симптомов: инфантильные спазмы, изменения на электроэнцефалограмме (ЭЭГ) в виде гипсаритмии и задержка психомоторного развития. Однако диагноз может быть установлен и при наличии двух признаков из трех [2, 3, 10]. Инфантильные спазмы — быстрые сокращения мышц продолжительностью 1–2 с, занимающие по своей скорости промежуточное положение между миоклоническими и тоническими приступами [1, 3]. Эпилептический спазм — более широкий термин, не тождественный синдрому Веста. Спазмы могут быть вне синдрома Веста в любом возрасте [1]. Согласно литературным данным, ряд зарубежных исследователей (Riikonen R., Yilmaz S., Nikolic D., Hamano S., Karvelas G., Mohamed B.P. et al.) старались выявить прогностически значимые факторы. Однако при анализе этих работ нами отмечены малая выборка пациентов, их неоднородность и преимущественно небольшой срок наблюдения. Кроме того, в данных работах имеются значительные различия в перечне наиболее важных прогностических признаков [4, 5, 7–9, 11–13, 15]. Таким образом, определение предикторов даст возможность прогнозировать исход синдрома Веста уже при первом клиническом осмотре пациента, определить выбор оптимальной терапии, сроки динамического наблюдения и улучшить результаты лечения.
Цель работы — определить прогностически значимые факторы исхода синдрома Веста, в том числе оценить зависимость исхода от этиологии заболевания, исследовать влияние эпилептических спазмов и других видов приступов на психомоторное развитие ребенка и развитие аутистикоподобного синдрома.
Материалы и методы
В исследование вошли 132 пациента, которые проходили стационарное лечение и наблюдались амбулаторно в условиях Нижегородской областной детской клинической больницы, Детской городской клинической больницы № 1 Нижнего Новгорода и Института детской и взрослой неврологии и эпилепсии имени святителя Луки Москвы с 2000 по 2018 г. Ранний анамнез у ряда пациентов оценивали по данным медицинской документации при анализе историй болезни и амбулаторных медицинских карт. У всех пациентов когда-либо регистрировали эпилептические спазмы. Среди вошедших в исследование детей 74 — мужского пола, 58 — женского. Таким образом, распределение детей по полу составило 1,28 : 1. Возраст детей на момент начала наблюдения составлял 5 мес. — 17 лет 11 мес. (средний 5 лет 9 мес. ± 4 года 5 мес.).
При оценке данных анамнеза всех пациентов разделили на три группы: 1-я группа — пациенты со структурной формой заболевания (60 пациентов; 45,5 %), 2-я группа — пациенты с генетической формой заболевания (39 детей; 29,5 %), 3-я группа — пациенты с невыясненной этиологией заболевания (33 ребенка; 25,0 %).
Возраст дебюта спазмов варьировал от 1-х суток жизни до 3 лет 2 мес. Ранний дебют эпилептических спазмов (до 3 мес.) наблюдали у 17 (12,9 %) детей, что не исключает наличие у ребенка других форм эпилепсии с последующей трансформацией в синдром Веста. Классический дебют (в возрасте 3–12 мес.) отмечали у 101 (76,5 %) ребенка, поздний дебют (в возрасте ≥12 мес.) — у 14 (10,6 %) детей. Пик дебюта спазмов приходится на возраст 4–6 мес.
У 125 (94,7 %) детей регистрировали серийные спазмы, у 7 (5,3 %) — одиночные. В 76 (57,6 %) случаях отмечали симметричные спазмы, в 56 (42,4 %) случаях — асимметричные. У 81,7 % детей выявляли флексорные, у 11,7 % — экстензорные и у 6,6 % — смешанные спазмы. В табл. 1 отражены дополнительные данные анамнеза пациентов сравниваемых групп.
Данные анамнеза пациентов
Читайте также: